Products
Customer Reviews
Resources
Support
Yeast is an invaluable organism in the biological research and industry. It is the simplest unicellular eukaryote. Its small genome size and the lower number of genes as well as the unicellularity make yeast one of the most important model system in cell biology and genetics, especially cell cycle regulation and signal transduction. Yeast converts carbohydrates to two products, carbon dioxide and alcohols. The former has been utilized for baking and the latter for brewing alcoholic beverages including beers and wine for thousands of years. Recently, the alcohol producing capability of yeast is applied to bioethanol production by using corn and sugar cane.
The current method of counting yeast is the ASBC (American Society of Brewing Chemists) method. In this protocol, yeast cells are stained with Methylene blue which is a cell membrane-impermeable dye. Dead yeast is stained in blue due to the compromised membrane integrity while live yeast is not. Because of the small size, yeast cells need to be observed in a high magnification (eg., 40X or higher objective lens). The magnification is inversely proportional to FOV (Field Of View). Researchers have to count both live and dead yeasts in the small square of the hemocytometer, move the microscope stage to cover a neighboring square and count live and dead yeasts in the next square. Counting continues until total 0.1µl counting volume is reached. In case of the central big tile of the hemocytometer, it corresponds to 25 small squares. It is tedious and painful (It can take up to 30 min!). The statistical significance of the ASBC method is low (25% error is typical) due to human errors, human interpretation and low counting volume (0.1µl). Moreover, in case of messy cultures (beer and wine brewing and bioethanol production), the manual counting is even difficult. Researchers need to distinguish yeast cells and non-cellular debris (hop in the beer brewing, grape in the wine brewing and corn mash in the bioethanol production) one particle at a time.
In contrast, it takes about 30 sec for LUNA-FL™ fluorescence cell counter to count yeast cells in the counting volume of 0.5µl which is five-fold more than the ASBC method.
Higher statistical significance can be obtained because no human error and no human interpretation are involved besides the larger counting volume. Users do not need to distinguish yeast cells from non-cellular debris (hops, grape and corn mash). In addition, cell viability can be measured by different criteria if two kind of dyes are used and their fluorescence excitation/emission wavelength are compatible with the optics of LUNA-FL™ fluorescence cell counter.
Fluorescein diacetate (FDA) is a fluorogenic cell viability probe. Due to its membrane-permeable nature, FDA can freely move in and out of the plasma membrane of diverse organisms from bacteria to mammalian cells. The internalized FDA is cleaved by intracellular esterases and converted to fluorescein. Fluorescein has the excitation maximum at 494nm and the emission maximum at 521nm, so it fits with the filter set for the green channel of conventional microscopes and LUNA-FLTM, automated fluorescence cell counter. The converted fluorescein carries one negative charge because of carboxylic acid located at the carbon numbered 3, so it cannot cross the plasma membrane any more. Its retention capability requires intact plasma membrane. Therefore, FDA staining measures the metabolic activity of esterase enzymes and the plasma membrane integrity. Overall working principle is summarized in Fig 1.
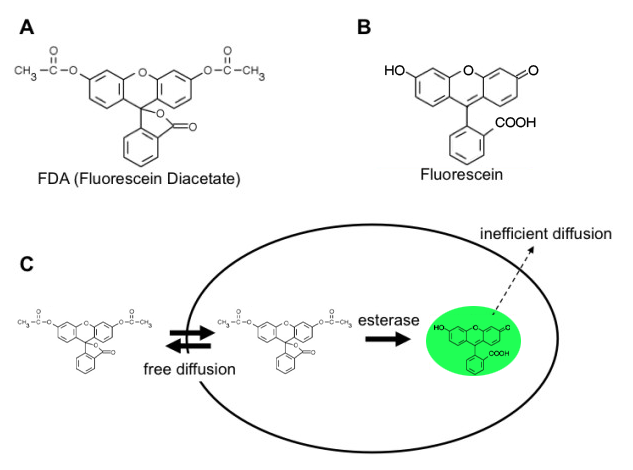
Figure 1. The working principle of yeast counting using FDA.
Saccharomyces Cerevisiae l Yeast culture medium (YPD) l LUNA-FL™, fluorescence cell counter l PhotonSlide™ for LUNA-FL™
Yeast viability kit (FDA and PI staining solution, Yeast dilution buffer, Fluorescence signal enhancer 1)
1. Overnight culture S. Cerevisiae in the YPD medium.
2. Dilute the confluent yeast at 1:100 and culture for additional 3h for mid-log phase cells (optional).
3. Dilute the yeast culture at 1:100 with the yeast dilution buffer (O/N culture) depending on the concentration of the yeast cells. Centrifuge, discard the supernatant and suspend the pellet with the yeast dilution buffer (the mid-log phase yeast). (optional) 4. Heat the yeast sample at 70℃ for 30 min to prepare 0% viability yeast cells. (optional)
5. Mix 1 µl of the fluorescence signal enhancer 1 and 17 µl of the yeast sample. (optional)
6. Incubate for 10 min at room temperature. (optional)
7. Add 1 µl of FDA and 1 µl of PI to the yeast sample.
8. Incubate for 10 min at room temperature.
9. Load 10 µl of the stained yeast sample on the counting slide.
10. Wait for about 1 min or until all yeast cells are immobile.
11. Turn on LUNA-FLTM and press the “Yeast Cell Counting (FDA/PI)”.
12. Perform the cell counting using the following protocol
As shown in Fig 2A, FDA can successfully stain all yeast cells when a yeast sample of 100% viability was tested. All yeast cells visible in the bright field image were counted and marked with the green circle (green/red circles represent the viable/dead cells automatically classified by the LUNA-FLTM). To see if FDA/PI staining can distinguish live and dead yeast cells, we intentionally killed yeast cells by heating at 70℃ for 30min. (0% viability sample) 50% viability sample was prepared by mixing 100% and 0% viability samples at a ratio of 1:1. As seen in Fig 2B and 2C, FDA/PI staining successfully distinguished live and dead yeasts even when they are mixed in a single vial. Like the 100% viability sample, all yeast cells were counted and labeled with green or red circle according to their health status. We next measured the precision of the viability measurement of LUNA-FL™ by using 0%, 25%, 50%, 75% and 100% viability samples. As seen in Fig 2D, the actual viability measured with LUNA-FLTM is significantly correlated with the theoretical viability with the coefficient of correlation, R2, of 0.9991. Taken together, the FDA/PI staining combined with LUNA-FLTM fluorescence counter can count yeast cells with the precise live/dead call.
Unlike AO/PI staining, in which two nucleic acid binding dyes bind instantly to genomic DNA, FDA staining make use of the enzymatic reactions for generating fluorescence. So it takes some time for the fluorescence intensity to reach a detectable level. To measure the optimal time required for the fluorescein detection, FDA was added to 100% live yeast cells, and the average fluorescence intensity of objects (yeast cells) was measured at 0, 5, 10, 15, 20 and 30min after the addition of the dye. As shown in Fig 3A, without the incubation time, the FDA fluorescence is not distinguishable from background (around 40). Within the first five minutes, the yeast cells rapidly gained the fluorescence signal. The rate of the fluorescein accumulation dropped significantly during the next five minutes and reached a plateau at 10min after the reaction. We decided to incubate yeast cells and FDA for 10min for optimal yeast counting.
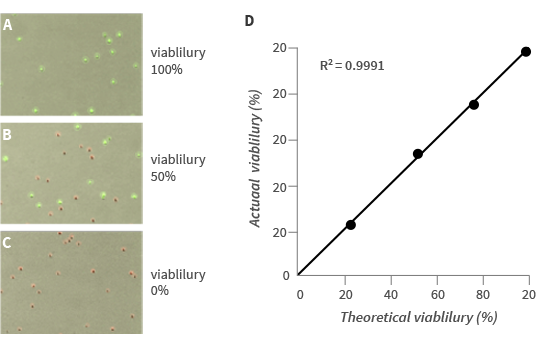
Figure 2. Automated yeast cell counting with the LUNA-FL™
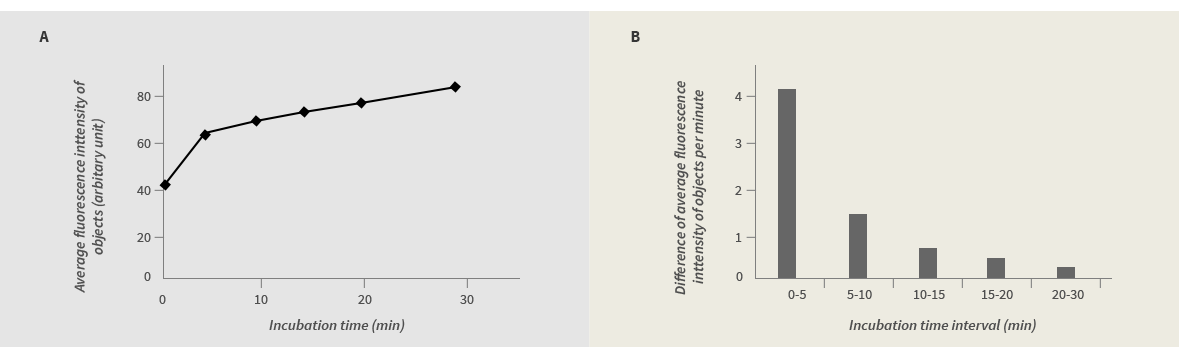
Figure 3. The kinetics of yeast staining by FDA
While FDA itself is not fluorescent as mentioned earlier, some cell culture media are auto-fluorescent (Fig 4A). This background fluorescence masks weak signals from some yeast cells. Moreover, a fresh YPD medium contains remnant esterase activities (Fig 4A/4B). If FDA is cleaved to fluorescein by the extracellular esterase activity, the fluorescein molecules can no longer enter yeast cells so that the background fluorescence becomes intense and, consequently, the object fluorescence becomes dimmer at the same time. The degree of the background fluorescence contributed by the esterase activity is much larger than YPD autofluorescence. The auto-fluorescence and the remnant esterase activity seem to originate from ingredients of the YPD medium, most likely yeast extract and peptone. Interestingly, the YPD medium used to culture yeast cells for overnight contains a lower level of the esterase activity compared to the fresh YPD medium. Seemingly, protein components containing the esterase activity in the YPD medium was absorbed by yeast cells and used as building blocks of yeast cells as they proliferate. In conclusion, some culture media contain auto-fluorescence and residual esterase activity. In this case, the culture medium should be diluted in an appropriate buffer, eg., yeast dilution buffer (Fig. 4C). Or, if the background fluorescence is still too bright, the whole yeast culture should be centrifuged, the resulting supernatant should be removed and its pellet should be re-suspended in the yeast dilution buffer.

Figure 4. The YPD medium is auto-fluorescent and contains esterase activity.
Depending on yeast strains and culture conditions, even flureoscein which has been activated by esterase enzymes can be leaky. The leakiness results in higher background and lower fluorescence intensity of yeast cells as seen in Fig 5A. A proprietary reagent (fluorescence signal enhancer 1) from Logos biosystems can help prevent the activated fluorescein from leaking out of yeast cells. As seen in Fig 5B, the background becomes darker and individual yeast cell becomes brighter because the fluorescein export was blocked by the fluorescence signal enhancer 1.
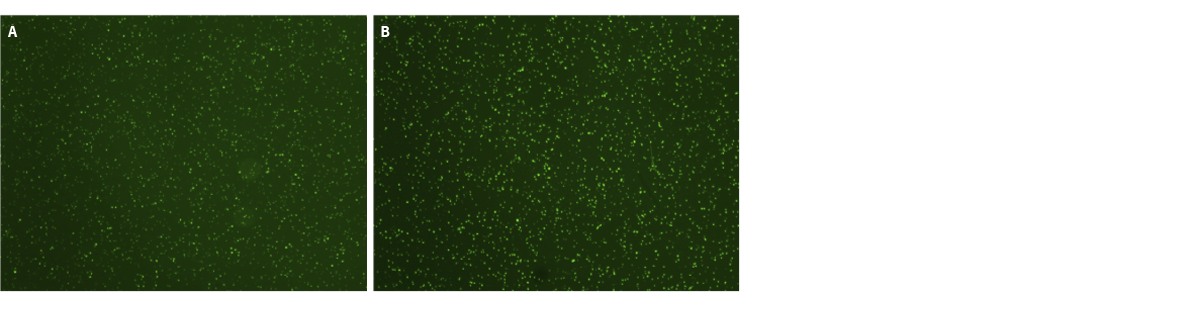
Figure 5. The fluorescence signal enhancer 1 dercreases the background fluorescene level and increases the fluorescence intensity of yeast cells.
LUNA-FL™ can calculate the total number and measure the viability of yeast with exceptional accuracy and precision within 30 sec when combined with FDA/PI, yeast dilution buffer and fluorescence signal enhancer 1. Because LUNA-FL™ can count yeast cells in messy culture as well as pure culture, a wide variety of academy, biotech/big pharma and alcohol industry can become beneficiaries.